Harmonized Standards Medical Device Software MDR IVDR
The new European regulations MDR (2017/745) and IVDR (7017/746) are based on a series of harmonized standards.
In this article we will delve into the harmonized standards for obtaining the CE marking of our medical or in vitro software-type product.
The following diagram shows a summary of the regulations that apply to medical and in vitro devices that include software:
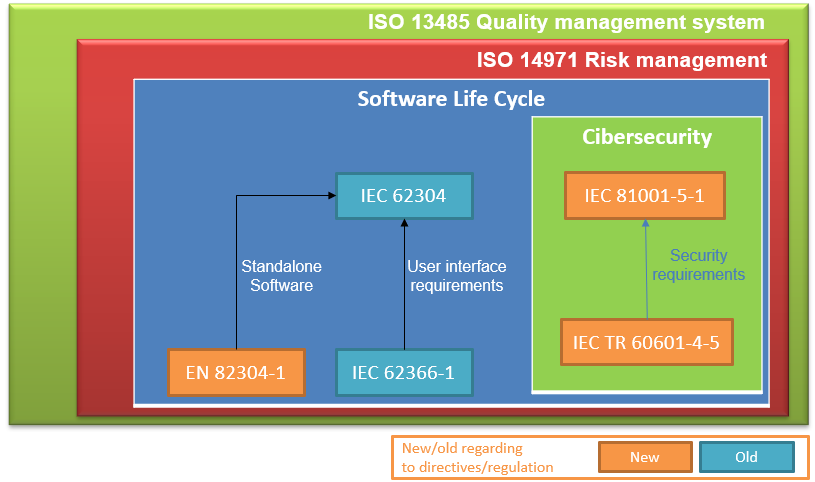
First of all, the regulation of the quality management system for medical devices is ISO 13485, the quality management standard that a company must comply with in order to obtain the CE marking for its software medical device.
Next, the standard immediately below and that 13485 refers to is the ISO 14971 risk management standard. Risk management is essential to be able to carry out the design and development tasks, and in order to then carry out a good validation of the product.
Under the quality framework of ISO13485 and following ISO14971 for the risk management of our product, we find the IEC 62304 standard, key to the design, development, validation, verification and maintenance of software, that is, it deals mainly with the software life cycle.
Another applicable standard that already existed in previous European directives was the usability standard IEC 62366-1. Applying this standard helps to specify user interface requirements that ensure that the risks associated with the usability of software-type medical devices are mitigated.
On the other hand, and as a novelty, there is the EN 82304-1 standard applicable to autonomous software.
And the biggest novelty in this new regulation is the incorporation of harmonized standards regarding cybersecurity. The IEC 81001-5-1 standard defines the secure software development life cycle, that is, it adds design, implementation, verification and validation activities to ensure that the software is designed to protect it from cybersecurity attacks. This regulation is complemented by the technical report IEC TR 60601-4-5, providing the security requirements that the software product must comply with.
IEC 62304: Software life cycle process
This regulation already appeared among the harmonized standards in the old European directives.
It is a functional safety standard that covers both the safe design and maintenance of medical device software. It provides processes, activities and tasks to ensure security.
When?
- The software is itself a medical device
- The software is an integrated part of the final medical device
The following image shows all the activities covered by this regulation:
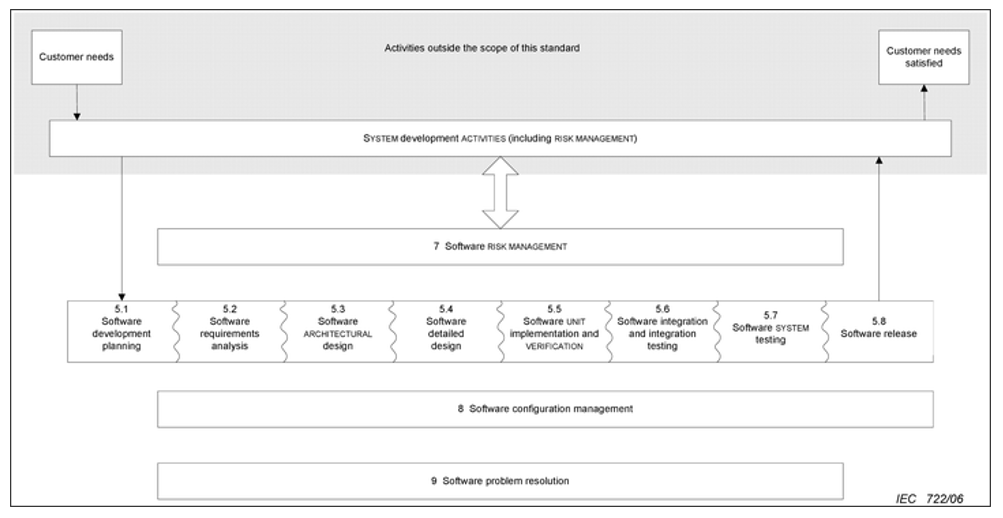
The activities in this regulation are divided according to the risk of the product. The 62304 has the following classification:
- A: No possible injury or damage to health
- B: Non-serious injury possible
- C: Death or serious injury is possible
Activities during software development
Next we focus on the activities of software development itself.
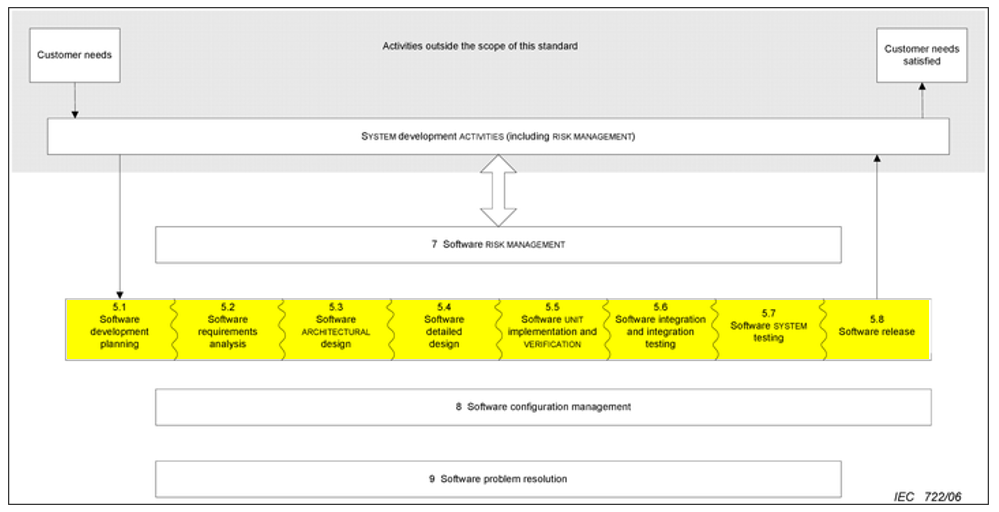
The following table summarizes the activities carried out according to the risk of the medical device:
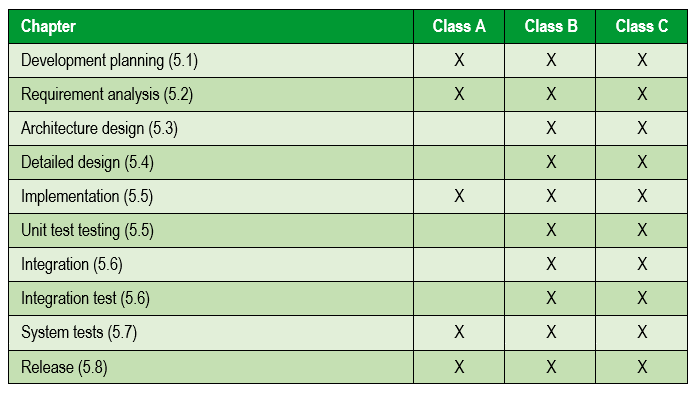
The following image summarizes the software development and validation activities included in the V model:
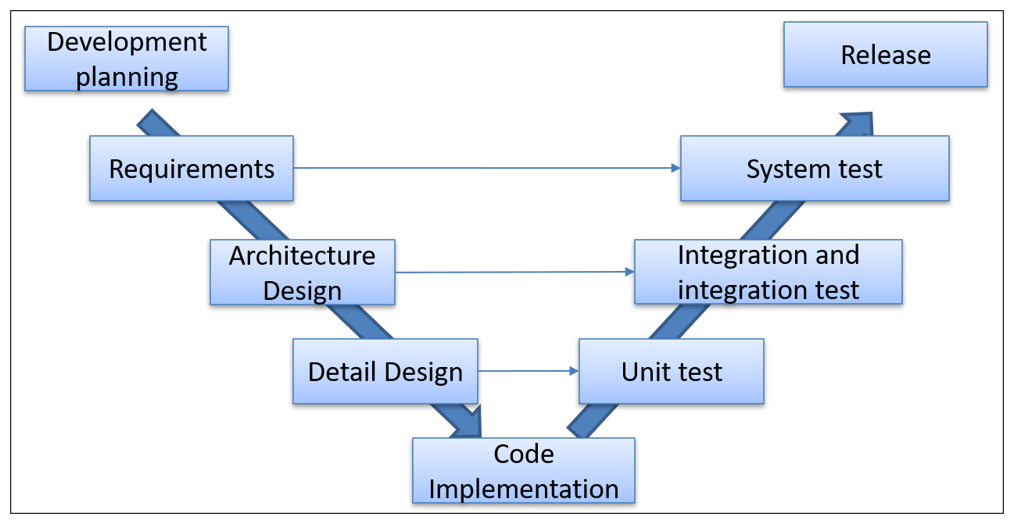
It can be seen that for a low level of risk the activities to be carried out are much lower than in the higher classes. There are also differences between risk classes B and C within each chapter of the standard.
EN 62366-1: Application of usability engineering to medical devices
This regulation is also one of those that was already among the harmonized standards in the old directives.
- Specifies a process for a manufacturer to analyse, specify, develop, and evaluate the usability of a medical device with regard to safety.
- This usability engineering process allows the manufacturer to assess and mitigate the risks associated with correct use and errors in use, that is, normal use.
- It can be used to identify risks, but it does not assess or mitigate the risks associated with abnormal use.
- This process is performed in parallel to the risk management process.
A summary of the scope of this regulation is presented in the following image:
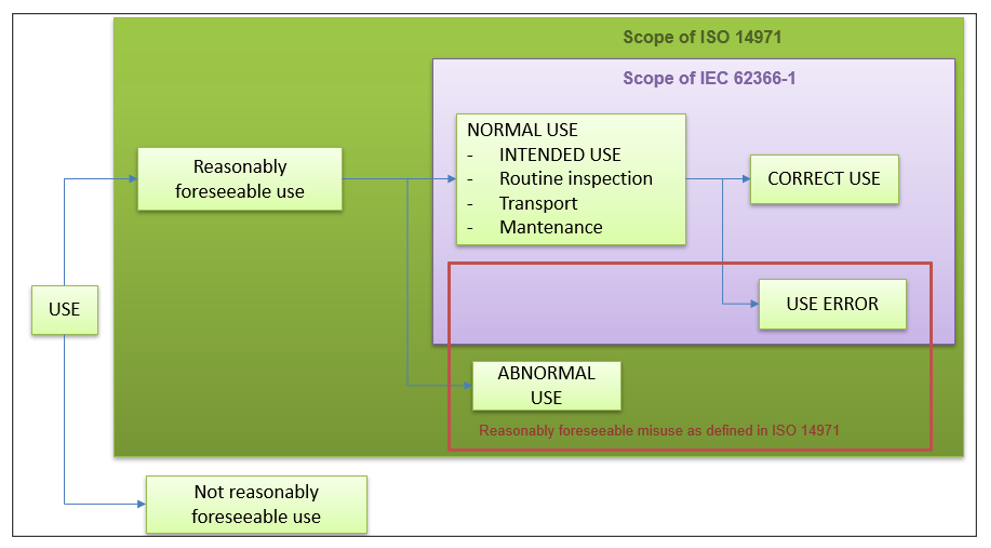
As can be seen, the scope of the usability regulation 62366-1 refers to the normal use of our software medical device.
The summary of activities to be carried out are the following:
- usability engineering plan
- Usability input data
- Preparation of use specification
- Analysis
- Design and formative evaluation
- Summative evaluation
EN 82304-1: Health software. Part 1: General requirements for product safety
The following regulations that we collect in this article because they appear as a harmonized standard for obtaining the CE marking on medical devices apply to the safety and protection of healthcare software products (the definition of healthcare software product includes medical devices) designed to operate on general computing platforms and intended to be released without dedicated hardware, and its primary focus is on requirements for manufacturers. It covers the entire life cycle, including the design, development, validation, installation, maintenance, and removal of healthcare software products.
Examples of medical devices to which this regulation would apply:
- Mobile apps
- Standard PC applications
- server applications
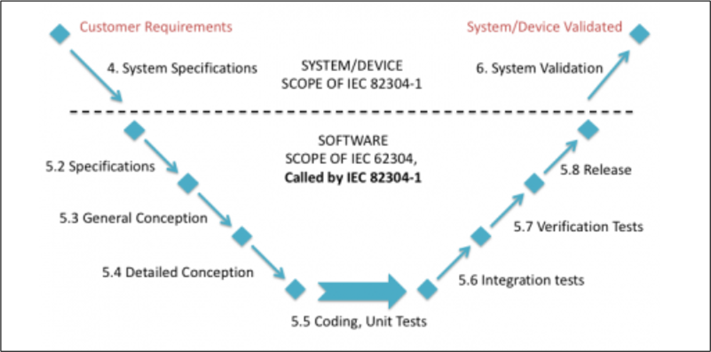
The following image shows the additional activities that are added to the 62304 regulation seen in previous sections:
IEC 81001-5-1: Safety and effectiveness of healthcare software and healthcare information technology systems. Part 5-1: Security. Activities in the product life cycle
The following standard is one of two harmonized standards dealing with cybersecurity in software medical devices.
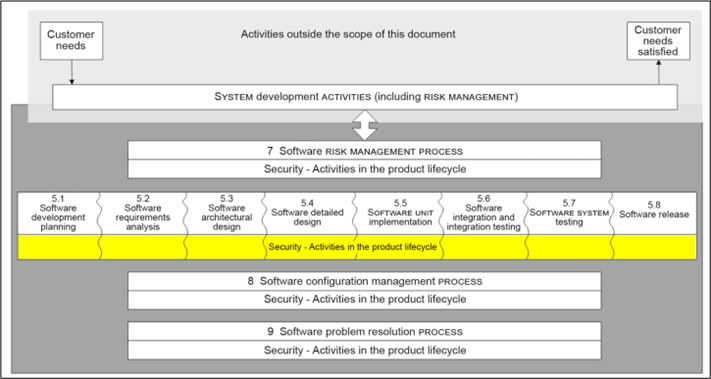
The IEC 81001-5-1 standard defines the life cycle requirements for the development and maintenance of healthcare software necessary to support compliance with IEC 62443-4-1, taking into account the specific needs of healthcare software.
The set of processes, activities and tasks establishes a common framework for the life cycle processes of insurance health software. The goal is to increase the cybersecurity of healthcare software by establishing certain activities and tasks in the healthcare software lifecycle processes and also by increasing the security of the software lifecycle processes themselves.
IEC TR 60601-4-5: Medical electrical equipment – Part 4-5: Guidance and interpretation – Safety-related technical security specifications.
And finally, we will talk about the technical report 60601-4-5, which provides detailed technical specifications for the security functions of medical devices used in medical IT networks.
Medical devices discussed in this document include:
- electrical medical equipment,
- electrical and medical systems
- medical device software
Note: Medical device software, although not within the scope of IEC 60601 (all chapters of this standard), you can also make use of this document.
This technical report is based on the seven basic requirements described in IEC TS 62443-1-1:2009 and provides specifications for different levels of medical device capability safety.
The specified security capabilities can be used to successfully integrate the device into defined security zones and conduits of a medical IT network with an appropriate medical IT network objective security level.
It is applicable to medical devices with external data interfaces used to capture sensitive data.
This technical report does not provide new validation and verification activities. It does provide the security requirements that our software medical device must have. The activities associated with these requirements were defined in regulation 81001-5-1.
References




Contact us
If you want to know more about the subject or have any other question, do not hesitate, contact us.
Request more information
Certificaciones

ISO 9001

ISO/IEC 27001

ENS-nivel medio

ISO 20000

UNE-EN ISO/IEC 17025
Síguenos
Aviso Legal | Política de Cookies | Contacto
© 2025 Software Quality Systems S.A. | SQS is a member company of Innovalia