Validación y Verificación de Software bajo MDR e IVDR
Los nuevos reglamentos europeas están generando mucho revuelo. Y ésto, ¿a qué se debe?
En este artículo os queremos transmitir qué dicen los nuevos reglamentos en cuanto a la validación y verificación del software en dispositivos médicos bajo la nueva regulación europea MDR/IVDR.
Contexto regulatorio europeo en dispositivos médicos
Normas armonizadas dispositivo médico e in vitro
Las directivas/regulaciones se apoyan en normativas/estándares para facilitar la aplicación de las mismas. La propia Comisión Europea se encarga de realizar la petición de creación o actualización de las mismas. A las normas realizadas por un organismo de normalización a raíz de una solicitud, se le denomina normas armonizadas. Las listas de normas armonizadas vigentes para cada directiva/reglamento se pueden encontrar en la web de la Comisión Europea.
Según la Guía azul:
“Si las referencias de las normas armonizadas se han publicado en el Diario Oficial de la Unión Europea, confieren una presunción de conformidad con los requisitos esenciales y otros requisitos legislativos que pretenden cubrir.”
Además,
“Si los fabricantes deciden no aplicar una norma armonizada, tienen la obligación de demostrar que sus productos cumplen los requisitos esenciales recurriendo a otros medios de su elección (por ejemplo, mediante cualquier especificación técnica existente, incluidas todas las demás normas disponibles).”
Como conclusión a todo lo anterior planteado, es buena práctica seguir las normas armonizadas que apliquen a nuestro producto. De esta manera:
- Será mas fácil cumplir con el reglamento ya que está mas detallado el alcance de los procesos a realizar que en la directiva/regulación
- Será más fácil de revisar la documentación técnica por un organismo notificado
- Al ser más fácil de revisar, será más fácil también conseguir la conformidad del producto.
Normas armonizadas productos software (año 2020)
Si buscamos la lista de normas armonizadas de las directivas anteriores a los nuevos reglamentos, relativas a software encontramos las siguientes:
- ISO 13485: Sistema de gestión de la calidad
- ISO 14971: Análisis de riesgos
- IEC 62304: Ciclo de vida del software
- IEC 62366-1: Productos sanitarios. Parte 1: Aplicación de la ingeniería de usabilidad a los productos sanitarios.
Normas armonizadas productos software (versión mayo 2022)
Vale, y ¿cuáles son las normas armonizadas que aplican actualmente?
- ISO 13485: Sistema de gestión de la calidad
- ISO 14971: Análisis de riesgos
Viendo las normas anteriores, el lector se preguntará cómo es posible que haya menos normas armonizadas que en las directivas anteriores.
¿Qué puede estar pasando? ¿Por qué hay menos normas armonizadas que antes? ¿Existe petición de actualizar las normas para convertirlas en armonizadas? La respuesta es SÍ.
Normas requeridas para ser armonizadas
En la siguiente lista aparece la lista de normas que aplicarían a dispositivos médicos que incluyan software:
- IEC 62304: Ciclo de vida del software
- IEC 62366-1: Usabilidad
- EN 82304-1: Software sanitario. Parte 1: requisitos generales para la seguridad de los productos
- IEC 81001-5-1: Seguridad y eficacia del software sanitario y de los sistemas de tecnología de la información sanitarios. Parte 5-1: Seguridad. Actividades en el ciclo de vida del producto
Actualmente la fecha máxima para adoptar estar normativas en los productos es mayo de 2024. Sin embargo, es probable que esta fecha se vea emplazada ya que se ha extendido el periodo de transición de la aplicación del nuevo reglamento.
Estrategia a seguir
Podríamos elegir entre una de estas opciones:
- ¿Esperar a que actualicen las normas y aparezcan en el listado de normas armonizadas?
- ¿Ir aplicando las versiones vigentes de las normas?
La elección depende mucho del contexto de cada empresa. Desde luego, una norma no se aplica ni interioriza en 2 días. Hay que tener en cuenta el tamaño del equipo de producto de la empresa para entender el tiempo que pueden llevar estas implementaciones. ¿Son temáticas novedosas o ya existe por ejemplo un departamento de ciberseguridad?
La sugerencia que damos desde SQS es ir aplicando las últimas versiones de las normas y en el momento que aparezcan actualizadas, revisar los cambios y aplicarlos.
Un punto positivo es que las normativas de ciberseguridad, que puede resultar el tema mas novedoso en este nuevo reglamento, ya están publicadas, aunque aun no aparezcan en la lista oficial de normas armonizadas.
Validación y verificación de software nuevo reglamento europeo
Definiciones validación y verificación
Suele haber confusión entre la diferencia entre los términos de validación y el de verificación.
Os traemos aquí las dos definiciones:
- Definición de Verificación: Proceso de examen seguido de un juicio basado en pruebas que demuestren que los elementos de salida (procesos, documentación, software o aplicaciones) de una fase de desarrollo específica cumplen con los requisitos de dicha fase en lo que se refiere a compleción, corrección y coherencia.
Ejemplo: revisión de documentos de diseño, pruebas unitarias
- Definición de Validación: proceso de análisis seguido de un juicio basado en pruebas para determinar si un elemento (por ejemplo, un proceso, documentación, software o una aplicación) responde a las necesidades de un usuario, en particular en lo que se refiere a seguridad y calidad, haciendo énfasis en la idoneidad de su funcionamiento de acuerdo con su objetivo en su entorno previsto.
Ejemplo: pruebas de sistema
Punto de partida
En los casos en los que ya tengamos un producto software funcionando y nos estemos planteando el sacar el marcado CE, las siguientes preguntas son las primeras que hay que contestar:
- ¿Tenemos claro que nuestro software es un dispositivo médico?
Para contestar de forma adecuada esta pregunta es necesario definir la intención de uso de nuestro producto.
Una vez hemos definido la intención de uso y, por tanto, podemos afirmar que nuestro software es un dispositivo médico podemos pasar a realizarnos la siguiente pregunta:
- ¿Tiene establecido mi producto la clase en función del riesgo?
Según si el producto es un dispositivo médico o in vitro se debe establecer. Dentro de los reglamentos existen reglas específicas para poder realizar esta clasificación de forma correcta. Existen guías específicas MDCG para abordarlo incluso debido a su complejidad a veces.
Teniendo los dos puntos anteriores resueltos, podemos pasar a hablar de las actividades de validación y verificación necesarias para cumplir con la nueva regulación de dispositivos médicos europea.
Actividades de Verificación y Validación nuevo reglamento
Las actividades en esta normativa se dividen en función del riesgo del producto. La 62304 tiene la siguiente clasificación:
- A: No es posible lesión o daño para la salud
- B: Es posible lesión no-seria
- C: Es posible muerte o lesión seria
En este artículo nos centramos en la validación y la verificación. La gestión de riesgos es muy importante pero está fuera del alcance de la validación. A partir de unos riesgos definidos se definen requisitos para mitigar los riesgos y esos requisitos son validados junto con el resto de requisitos.
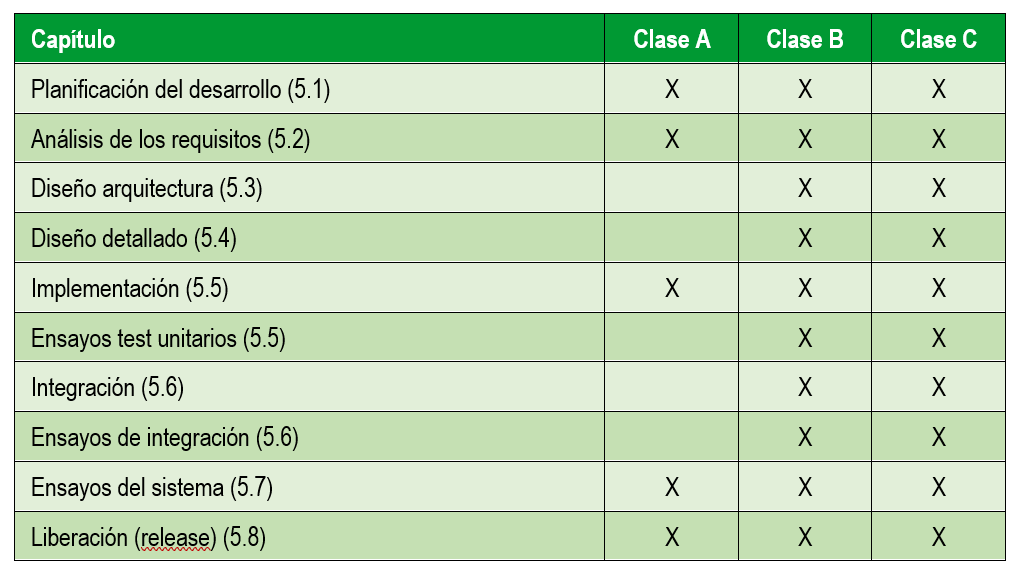
A continuación nos centramos en las actividades del desarrollo del software en sí. En la siguiente tabla se resumen las actividades recogidas en la normativa 62304 y que se realizan en función del riesgo del dispositivo médico:
En la siguiente imagen se muestran todas las actividades de desarrollo, validación y verificación de software recogidas en el modelo en V:
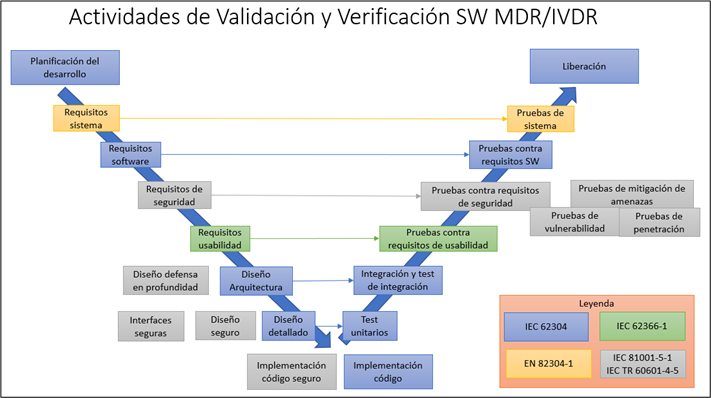
Y como resultado de todas actividades se realiza un informe de validación y verificación de software que debe contener los resultados de todas las actividades anteriores y se recoge en los siguientes apartados:
Actividades de validación de software producto sanitario MDR/IVDR
Entre las actividades de validación que hay que realizar, que incluyen tanto especificación como ejecución y realización de informe de pruebas se encuentran las siguientes:
- Pruebas para los requisitos software (62304)
- Pruebas para los requisitos de usabilidad (62366-1)
- Pruebas para los requisitos de sistema (82304-1)
- Pruebas para los requisitos de seguridad (81001-5-1)
- Pruebas de mitigación de amenazas (81001-5-1)
- Pruebas de vulnerabilidad (81001-5-1)
- Pruebas de penetración (81001-5-1)
- Repetición de pruebas del sistema software (5.7.4 y 5.7.5)
Actividades de verificación de software producto sanitario
Resultado de la revisión de la documentación:
- Requisitos sistema (82304-1)
- Requisitos software (62304 – apartado 5.2.6)
- Requisitos usabilidad (62366-1)
- Requisitos de seguridad (81001-5-1)
- Arquitectura del software (62304 – apartado 5.3.6)
- Diseño defensa en profundidad (81001-5-1)
- Diseño seguro (81001-5-1)
- Diseño detallado (62304 – apartado 5.4.4)
- Interfaces seguras (81001-5-1)
- Integración software (62304 – apartado 5.6.2)
- Procedimientos de pruebas de integración (62304 – apartado 5.6.5)
Resultado de pruebas:
- Pruebas unitarias (62304 – apartado 5.5.5)
- Pruebas de integración (62304 – apartado 5.6.2)
- Pruebas de regresión (62304 – apartados 5.6.6 y 5.6.7)
Los errores encontrados durante las pruebas se suelen recoger en los siguientes informes:
- Informe de errores (pendientes de solucionar)
- Informe de errores residuales
En el informe de validación y verificación software se incluye además una matriz de trazabilidad donde se enlazan los requisitos con los resultados de las ejecuciones de casos de prueba. Con ella se demuestra que no existe ningún requisito sin probar. Si existe alguna ejecución con resultado fallido se verá también reflejado en esta matriz.
Resumen
En este momento debemos:
- Revisar periódicamente la lista de normas armonizadas
- Revisar la publicación de las normas que aun no están actualizadas
Y mientras tanto:
- Diseñar y validar nuestro producto sanitario -> basado en el riesgo (safety)
- Diseñar y validar nuestro producto sanitario -> basado en la ciberseguridad (security)
Conclusión: ¿Qué es lo novedoso del nuevo reglamento que afecta al diseño del producto sanitario software?
Lo mas novedoso sin lugar a dudas es la implementación de medidas de ciberseguridad.
Contacta con un experto
Si quieres saber más del tema o tienes cualquier otro tipo de consulta, no lo dudes, ponte en contacto con nosotros.
Solicita más información
Certificaciones

ISO 9001

ISO/IEC 27001

ENS-nivel medio

ISO 20000

UNE-EN ISO/IEC 17025
Síguenos
Aviso Legal | Política de Cookies | Contacto
© 2025 Software Quality Systems S.A. | SQS is a member company of Innovalia